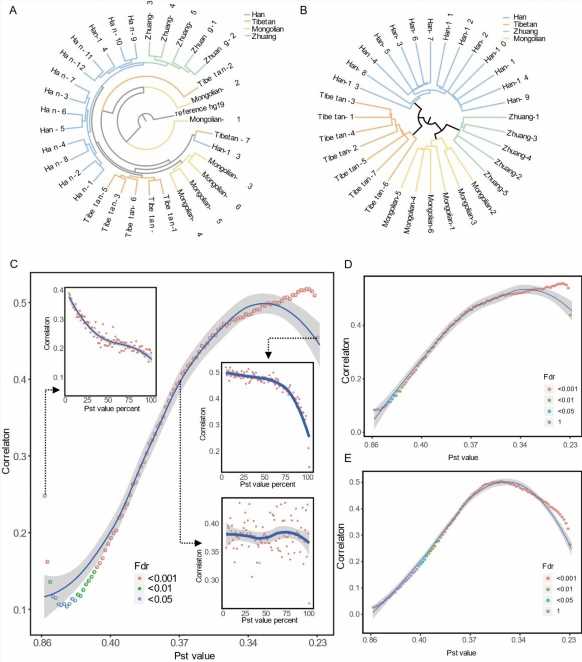
 The genetic structure of the population based on population SNPs. (B) The epigenetic structure of the population based on ethnic-specific DNA methylation sites. (C) The distribution curve of correlation values estimated between overall genetic distances and CpG methylation Manhattan distances of CpGs sorted with the values of Pst, the parameter to estimate phenotypic differentiation between populations. (D-E) The correlation curves based on the between-ethnic (D) or within-ethnic (E) comparison of genetic distances with CpG methylation Manhattan distances. Credit: Science China Press")
A paper titled “Genome-wide DNA methylation landscape of four Chinese populations and epigenetic variation linked to Tibetan high-altitude adaptation” has been published online in Science China Life Sciences.
This study was led by Prof. Wen Wang (Northwestern Polytechnical University), Prof. Shuhua Xu (Fudan University), Prof. Zhengsheng Sun (Chinese Academy of Sciences), and associate Prof. Peng Tian (Northwest A&F University). Using the recently developed double-strand bisulfite sequencing (DSBS) from Sun’s lab, this study revealed both genetic and DNA methylation variation in four Chinese ethnic groups, and investigated the potential difference and association of the two mechanisms in the population. Based on the comparison of Tibetans with other lowland individuals, the researchers proposed potential function of epigenetic regulation in Tibetans’ adaptation to Qinghai-Tibet Plateau.
DNA methylation is one of the most important epigenetic marks in eukaryotic genome, and plays an important role in multiple cellular processes. Ongoing research has shown the variation of DNA methylation among different human populations, and how it is associated with both genetic and environmental factors.
China is the most populous country in the world, consisting of 56 recognized ethnic groups. Geographical distribution and habitat differences are obvious among some ethnic populations. The most typical example is Tibetans’ adaptation to the Qinghai-Tibet Plateau, which is low in oxygen while high in UV radiation. However, until the publication of this work, few studies have analyzed the variation of DNA methylation among different ethnic groups in China, not to mention the role of epigenetic regulation in Tibetans’ adaptation to high-altitude.
In this work, the authors collected blood samples from 32 participants from among four Chinese ethnic groups including Chinese Han, Tibetan, Mongolian, and Zhuang. They carried out DSBS, a recently developed high-throughput sequencing technology that can accurately identify both single-nucleotide variants and DNA methylation simultaneously at a single-base resolution by using one data set.
The results of this study suggest that DNA methylation-based epigenetic structures differ greatly, with genetic structures that clearly distinguish the four ethnic groups. Only a small part of the DNA methylation sites shows ethnic difference and could separate the four groups. Surprisingly, after excluding the influence of within-ethnic and between-ethnic difference, the researchers found non-ethnic-specific DNA methylation variations correlated more significantly to global genetic divergence than these ethnic-specific DNA methylation structures, suggesting ethnic-specific DNA methylation variations were determined more by environmental factors other than genetic diversifications.
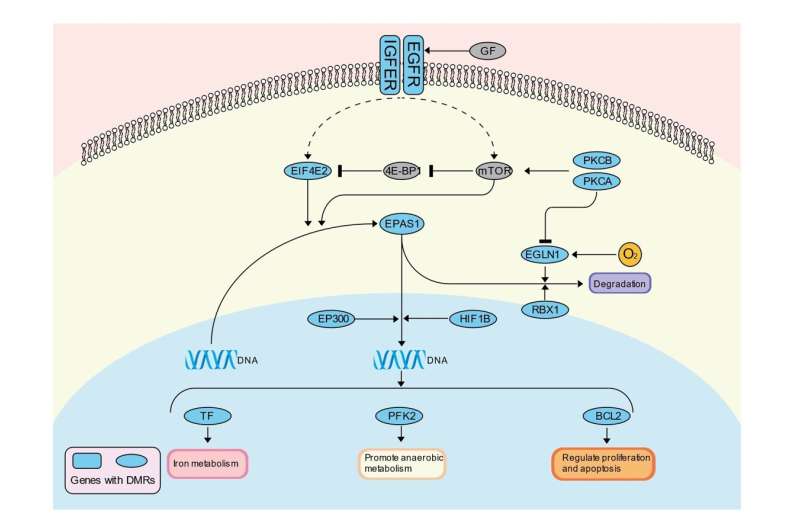
Interestingly, the data revealed that DNA methylation differences between Tibetan and other lowland individuals were enriched around high-altitude adaptation related genes including EPAS1 and EGLN1, which were critical selection signals in Tibetans’ adaptation to highlands, suggesting that DNA methylation alteration plays an important role in high-altitude adaptation.
Additionally, the researchers also discussed the accuracy of nanopore sequencing in human epigenomic studies, and explored the reliability and feasibility of this new technology at DNA methylation identification as compared to DSBS.
This work provides the first batch of epigenetic comparison maps for some important Chinese populations and the first evidence of the association of epigenetic changes with Tibetans’ high-altitude adaptation.
More information:
Zeshan Lin et al, Genome-wide DNA methylation landscape of four Chinese populations and epigenetic variation linked to Tibetan high-altitude adaptation, Science China Life Sciences (2023). DOI: 10.1007/s11427-022-2284-8
Journal information:
Science China Life Sciences
Source: Read Full Article