. DOI: 10.1038/s41588-023-01608-3")
Each gene in the human genome has two copies. When researchers detect two mutations within a particular gene in a patient’s genome, it can be difficult or expensive to determine if those two mutations are present in the same copy of the gene (“in cis”) or different copies of the gene (“in trans”).
A team led by investigators at Massachusetts General Hospital (MGH) and the Broad Institute of MIT and Harvard recently developed a strategy for inferring which of these phases is present for rare variant pairs within genes.
As reported in Nature Genetics, the work will be helpful for interpreting findings from clinical genetic testing—especially for recessive diseases, which arise when both copies of a gene are impacted by a damaging genetic variant.
For the study, researchers analyzed sequencing data of the expressed genes—or the protein coding regions of the genome—from 125,748 individuals from the Genome Aggregation Database (gnomAD), a large international public open-access human genome resource.
The team applied a statistical method called an expectation-maximization algorithm to the genetic data from gnomAD to estimate whether a pair of rare variants are seen in cis or in trans.
“Our method to estimate the phase of rare variants was 96% accurate in two independent datasets, including a set of patients with recessive Mendelian conditions,” says senior author Kaitlin E. Samocha, Ph.D., an Assistant Investigator in the Center for Genomic Medicine at MGH and an Associated Scientist at the Broad Institute of MIT and Harvard.
“The accuracy of our approach remained high even for very rare variants and across genetic ancestry groups.”
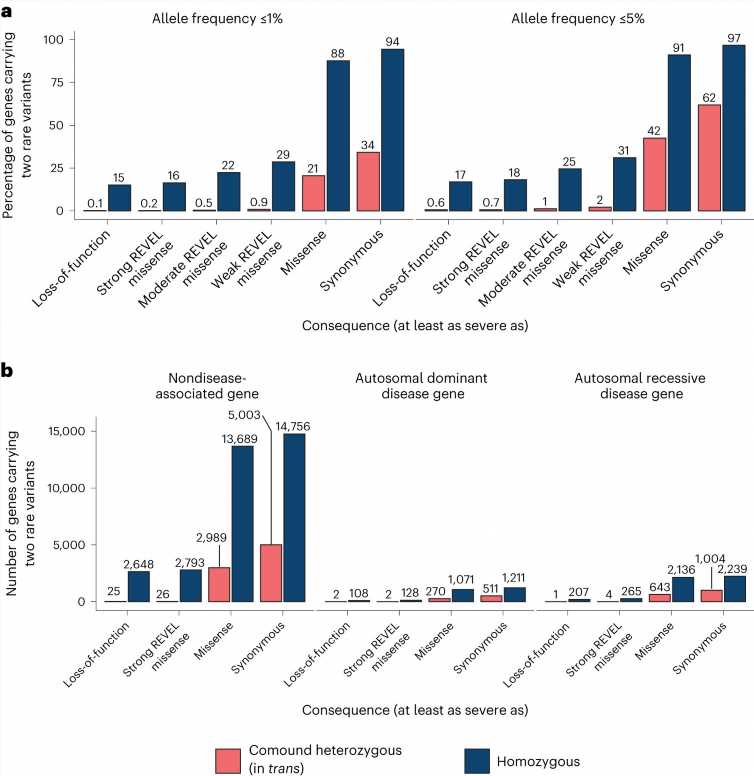
Additionally, the investigators found that only a small number of genes were impacted by loss-of-function variants predicted to be in trans, which would be predicted to lead to the complete loss of that protein.
In most individuals, if two rare loss-of-function variants were found in the same gene, the variant pair was in cis. Therefore, when a pair of rare loss-of-function variants is observed in the same gene in an individual in the general population, it is more likely that these variants are carried on the same copy of the gene rather than on different copies.
“We have publicly released phasing predictions for over five billion pairs of rare variants seen in the gnomAD dataset, as well as our counts per gene of variant pairs predicted to be in trans, at gnomad.broadinstitute.org,” says Samocha.
Although this work focused on estimating the phase of rare coding variants in expressed genes, Samocha and her colleagues hope to incorporate noncoding and other variant types in their phasing estimates.
“Additionally, as more genome sequencing data become available, we will evaluate how our approach compares with more sophisticated phasing algorithms,” she says. “Finally, we will seek out more evaluations of the utility of our approach in a clinical genetic setting.”
More information:
Michael H. Guo et al, Inferring compound heterozygosity from large-scale exome sequencing data, Nature Genetics (2023). DOI: 10.1038/s41588-023-01608-3
Journal information:
Nature Genetics
Source: Read Full Article