A recent study posted to the bioRxiv* preprint server investigated the active genetic evolution of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) main protease (Mpro), its emerging mutations, and the existence of antiviral resistance against Mpro inhibitors like paxlovid.
The emergence of novel SARS-CoV-2 variants has necessitated the development of therapeutic approaches against coronavirus disease 2019 (COVID-19). However, the potential antiviral resistance emerging prior to, during, and after the introduction of such therapies needs extensive research.
 Study: Genetic surveillance of SARS-CoV-2 Mpro reveals high sequence and structural conservation prior to the introduction of protease inhibitor Paxlovid. Image Credit NIAID
Study: Genetic surveillance of SARS-CoV-2 Mpro reveals high sequence and structural conservation prior to the introduction of protease inhibitor Paxlovid. Image Credit NIAID
About the study
In the present study, researchers monitored the genetic changes occurring in SARS-CoV-2 Mpro and the development of potential mutations that could evade the human immune response. They also assessed the efficiency of Mpro as a COVID-19 drug target and provided an understanding of the diversity of Mpro prior to the introduction of paxlovid.
The team investigated the conservation of Mpro proteins across the Coronaviridae family by examining the active sites found on the α-CoVs, β-CoVs, and γ-CoVs structures. The active sites on the amino acid sequences on these CoVs and the differences in structural conformations of multiple Mpro enzymes were assessed by comparing the same to selected Protein Data Bank (PDB) structures. The authors selected 26 amino acids as active site residues.
Amino acid changes in Mpro were monitored using an in-house annotation pipeline that enabled the team to regularly retrieve and annotate the Mpro sequences of different SARS-CoV-2 genomes. The study also investigated the mutation frequency of more than 4.8 million Mpro isolates at the Mpro substrate cleavage sites and the sequence conservation of neighboring amino acid residues along with the open reading frame 1 ab (ORF1ab).
Results
The study results showed that the sequence homology comparison of the SARS-CoV-2 Mpro amino acid residues across different CoVs highlighted catalytic site residues and SARS-CoV-2 spike-1 (S1) pocket residues were identical in the Mpro of all the CoV sequences. Furthermore, amino acids residues present at S2 and S4 pockets were also more diverse than the residues at S1. Moreover, the residues in S4 showed even more remarkable diversity as compared to the residues in S2. Notably, while S2 and S4 were not entirely conserved across the proteases of different CoVs, these pockets had significantly identical amino acid sequences.
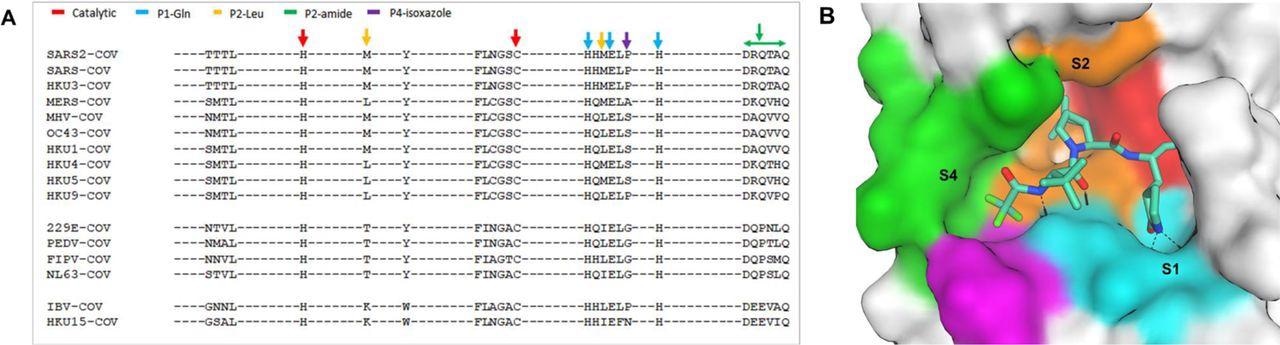
Active site conservation of CoV main proteases.
A) Sequence alignment of the 26 binding site amino acids. The key amino acids are indicated by color-coded arrows based on their interaction with the inhibitor, nirmatrelvir. B) SARS-CoV-2 Mpro binding pocket of nirmatrelvir. The pocket surface is colored based on the inhibitor’s interaction shown in panel A.
Furthermore, the study findings also showed that SARS-CoV and SARS-CoV-2 shared 100% identity and similarity at the active sites. Overall, the Mpro sequence and structure were highly conserved among the studied CoVs and the nirmatrelvir binding pockets.
Almost 84% of Mpro isolates shared the same amino acid residue sequence as that of the PDB reference isolate, including approximately 14K unique nucleotide alleles and around 4,800 protein variants. Additionally, the non-synonymous mutation rate (substitution/residue/year) was found to be lower for Mpro than for ribonucleic acid (RNA)-dependent RNA polymerase (RdRp) and over 10-times lower for the S protein. Notably, the study observed the first rise in the non-synonymous mutation rate in the S gene between November 2020 and December 2020, which overlapped with the emergence of the SARS-CoV-2 Alpha and Beta variants of concern (VOCs).
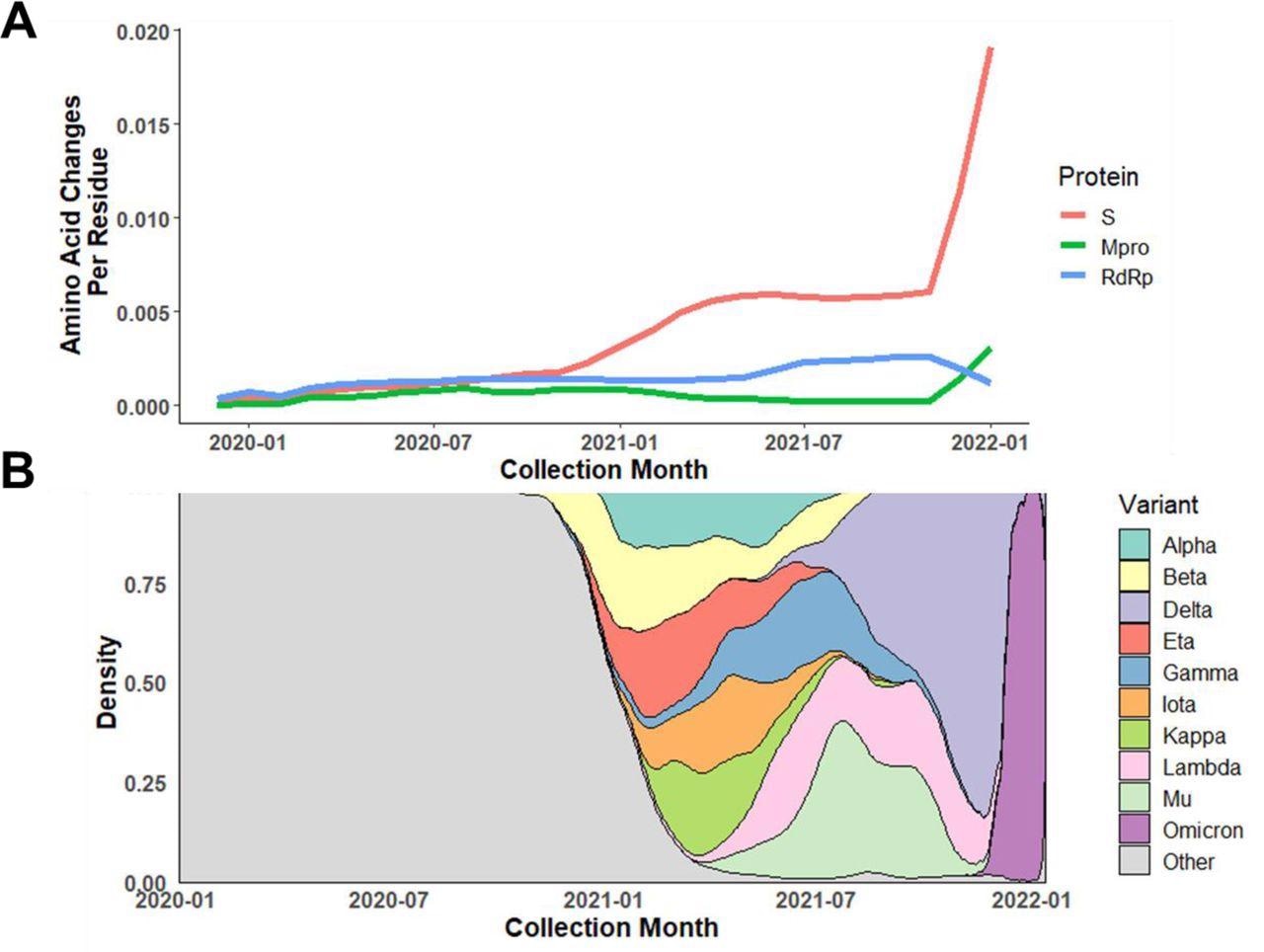
Dynamic change in amino acid mutation rate of Mpro compared to S protein and RdRp.
A) Average amino acid changes per residue in Mpro, S protein, and RdRp among isolates collected from January 2020 through January 2022. B) Relative distribution of VOC/VOIs based on collection date. The rapid rise in amino acid changes found in S protein and Mpro near the end of 2021 corresponds to the emergence and takeover of Omicron.
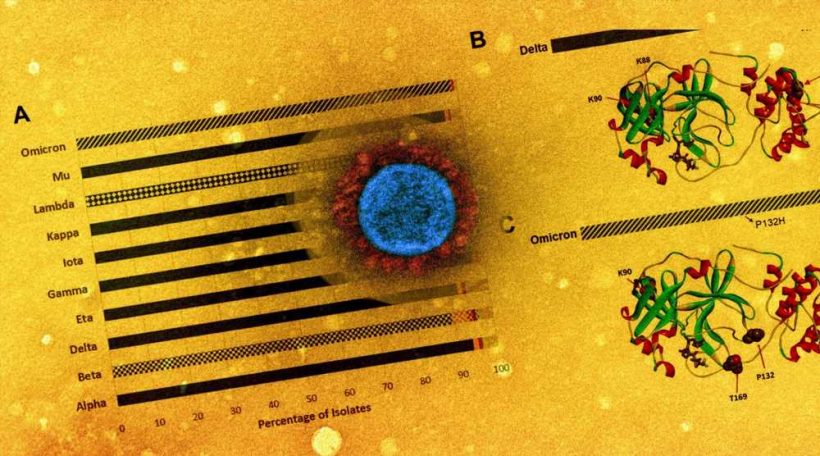
The examination of the SARS-CoV-2 genomes showed that the P132H was the most prevalent mutation, detected in more than 98% of the Omicron isolates, while the G15S and K90R mutations were predominant in the SARS-CoV-2 Lambda and Beta isolates, respectively. Moreover, nine key residues, namely histidine-41 (His41), methionine-49 (Met49), glycine-143 (Gly143), and cysteine-145 (Cys145), His163, His164, Met165, glutamic acid-166 (Glu166), and glutamine-189 (Gln189) were identified in the co-crystal structure of the Mpro-nirmatrelvir complex. Among these, His41 and Cys145 were catalytic residues, while the rest directly interacted with nirmatrelvir.

Mpro mutation breakdown at nirmatrelvir contact and catalytic residues.
A) Mutations identified at residues directly interacting with nirmatrelvir and/or substrate peptide. B) Three-dimensional structural model of Mpro (PDB ID 7RFS.pdb), with residues from panel A highlighted in “stick” representation and shown in individual colors. Protein backbone is shown in ribbon representation.
A total of 445 unique amino acid alterations were identified within the five residues of the Mpro substrate cleavage sites. Notably, while the P1 Gln residue was the most conserved amino acid found on the reference sequence, P2 and P1 positions showed fewer mutations in all the examined isolates.
Conclusion
The study findings showed that the structural conservation and the genetic stability of SARS-CoV-2 Mpro indicated the lack of any pre-existing resistance to nirmatrelvir. The researchers believed this study presents an established system that allows genetic surveillance using real-world genomic data. The constant emergence of SARS-CoV-2 variants and their evolution under the various selective pressures will prove critical in future studies against COVID-19.
*Important notice
bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
- Genetic surveillance of SARS-CoV-2 Mpro reveals high sequence and structural conservation prior to the introduction of protease inhibitor Paxlovid. Jonathan T. Lee, Qingyi Yang, Alexey Gribenko, B. Scott Perrin Jr., Yuao Zhu, Rhonda Cardin, Paul A. Liberator, Annaliesa S. Anderson, Li Hao, bioRxiv 2022, DOI: https://doi.org/10.1101/2022.03.29.486331, https://www.biorxiv.org/content/10.1101/2022.03.29.486331v1
Posted in: Medical Research News | Disease/Infection News | Pharmaceutical News
Tags: Amino Acid, Coronavirus, Coronavirus Disease COVID-19, covid-19, Cysteine, Evolution, Frequency, Gene, Genetic, Genomic, Glutamic Acid, Glutamine, Glycine, Histidine, Immune Response, Methionine, Mutation, Nucleotide, Omicron, Polymerase, Protein, Research, Respiratory, Ribonucleic Acid, RNA, SARS, SARS-CoV-2, Severe Acute Respiratory, Severe Acute Respiratory Syndrome, Syndrome

Written by
Bhavana Kunkalikar
Bhavana Kunkalikar is a medical writer based in Goa, India. Her academic background is in Pharmaceutical sciences and she holds a Bachelor's degree in Pharmacy. Her educational background allowed her to foster an interest in anatomical and physiological sciences. Her college project work based on ‘The manifestations and causes of sickle cell anemia’ formed the stepping stone to a life-long fascination with human pathophysiology.
Source: Read Full Article