")
An important aspect of drug discovery involves determining how well a drug binds to its target (protein) molecule. Typically, this step involves aligning a 3D structure of a drug and its target protein at various configurations in a process called “docking.” Preferred binding sites are then discovered by running docking simulations repeatedly with multiple drug candidates for a particular target molecule. Typically, owing to the vast number of likely candidates for potential drug-target interactions, deep learning models are used to carry out such simulations. However, a problem with using such models is the difficulty in interpreting their predictions. While deep learning certainly makes for a speedy drug discovery, it is little more than a black box. Additionally, docking simulations cannot be used to develop drugs for novel targets that have no known 3D complex with an interacting drug.
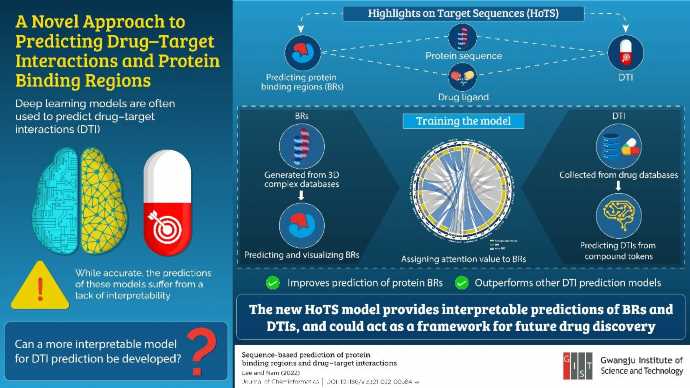
In a recent study published in the Journal of Cheminformatics, Associate professor Hojung Nam and her Ph.D. student Ingoo Lee from the Gwangju Institute of Science and Technology in Korea developed an new deep learning model called “Highlights on Target Sequences” (HoTS) for predicting the binding between a drug and a target molecule. The new model not only makes better predictions but, more interestingly, did so in an interpretable manner. Moreover, the model could predict drug-target interactions (DTIs) without the need for simulations or 3D structures.
How did the team achieve this feat? Professor Nam explains how their drug-target interaction prediction model works: “First, we explicitly teach the model which parts of a protein sequence will interact with the drug using prior knowledge. The trained model is then utilized to recognize and predict interactions between drugs and target proteins, giving better prediction performances. Using this, we built a model that can predict the target proteins’ binding regions and their interactions with drugs without a 3D-complex.”
Rather than dealing with the complete length of the protein sequence, the model could make predictions based only on the parts of the protein that are relevant to the DTI interaction. “We taught the model where to ‘focus’ to ensure that it can comprehend important sub-regions of proteins in predicting its interaction with candidate drugs,” elaborates Professor Nam. This, in turn, allowed the model to predict DTIs more accurately than existing models.
Source: Read Full Article