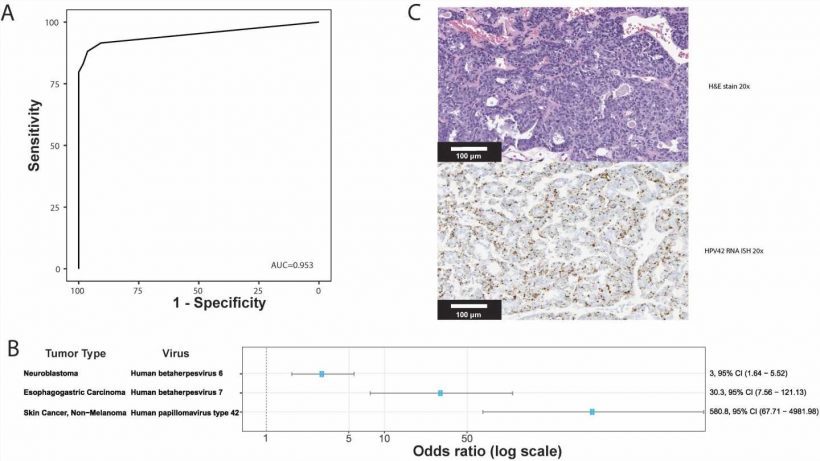
 curve showing performance of virus detection compared to isothermal amplification method. B: Forest plot showing enrichment of HHV6 in neuroblastoma, HHV7 in esophagogastric cancer, and HPV42 in non-melanoma skin cancer. Results are shown as an odds ratio comparing the enrichment for the virus in the specific tumor type compared to the other tumor types in the cohort. To the right of the figure, the odds ratio values and 95% confidence intervals are shown. C: The top panel shows a photomicrograph demonstrating the morphology of digital papillary adenocarcinoma by hematoxylin & eosin stain at 20X magnification, and the bottom panel shows photomicrograph of same tissue with RNA in situ hybridization custom designed to detect HPV42 demonstrating the localization of the virus to tumor cells. Credit: <i>Journal of Molecular Diagnostics</i>")
Researchers have developed a method to accurately detect viruses from clinical next-generation sequencing and describe novel associations between specific tumors and viruses that warrant further investigation. This information makes it more feasible to consider viral status in treatment protocols. Their findings appear in the Journal of Molecular Diagnostics.
Viruses commonly transfer from normal human cells to malignant cells in solid tumors. However, virus detection is limited in current clinical practice. Universal screening of viruses is not technically or fiscally viable because current standard of care techniques are uniplex, costly, and have challenging workflows.
Researchers developed a digital subtraction technique, which deletes human genome cells from sequencing analysis to identify the presence of viral DNA as a quality assurance (QA) process. This bioinformatic technique requires no additional sequencing, and virus detection can be achieved with minimal additional cost. Their results were comparable to standard clinical methods for tumor virus identification.
“We decided to look at tumor types that are commonly associated with a virus, and in almost all cases, the QA tool detected the virus we expected,” explained lead investigator Chad M. Vanderbilt, MD, Department of Pathology, and Department of Laboratory Medicine, Memorial Sloan Kettering Cancer Center, New York, NY, U.S.. “With this encouraging finding, we decided to refine the method as a microbiome detection pipeline and extend the analysis to see how well the method works for detecting clinically relevant viruses and discovering unexpected virus-tumor relationships.”
The study is the largest and most comprehensive study of human DNA virus detection in cancer. It used data gathered from January 2014 to October 2020 from 48,148 solid tumors sequenced by a US Food and Drug Administration–cleared tumor profiling assay for patients with advanced solid tumors. A BLAST (basic local alignment search tool) algorithm compared the non-human (unmapped) sequencing reads present in the tumors against all human viruses from the National Center for Biotechnology Information Virus database. Researchers cross-validated their results with multiple methods across tumor types and virus species and found their method has comparable sensitivity for detecting high risk human papilloma viruses (HPV) and Epstein Barre virus (EBV) to clinically validated in situ hybridization and amplification methods.
Investigators then extended the analysis to discover novel tumor-virus associations. Previously unreported associations between human herpes virus (HHV)6 in neuroblastoma and HHV7 in esophagogastric cancer were validated using an independent dataset. They also found a new association between HPV42 and digital papillary adenocarcinoma. In comparison to performing laborious single virus discovery assays, having access to data for discovery by data analysis alone allows resources to be dedicated to investigating the role that viruses might play in oncogenesis and for consideration of virus-informed therapies.
Source: Read Full Article