
A research team from Kumamoto University, Japan has developed an animal model that reproduces motor dysfunction and cerebellar neurodegeneration similar to that in spinocerebellar ataxia (SCA) by inhibiting chaperone-mediated autophagy (CMA) in cerebellar neurons. Since CMA activity is reduced in cells expressing SCA causing proteins, CMA is expected to become a new therapeutic target for SCA—a disease that currently has no basic treatment.
SCA is an inherited, intractable neurological disorder caused by several genes. It causes atrophy and neurodegeneration of the cerebellum and is characterized by progressive motor dysfunction like staggering and dysarthria (a brain disorder that results in a loss of control the muscles used in speech). It is classified into 48 different types according to the causative genes, and the common mechanisms that lead to cerebellar atrophy and ataxia in SCA are not yet understood.
The cells of living organisms have a mechanism called autophagy that maintains homeostasis by breaking down intracellular components, such unnecessary proteins, and eliminating pathogenic microorganisms. Chaperone-mediated autophagy (CMA), a type of autophagy, transports intracellular components to lysosomes for degradation using Hsc70, a molecular chaperone, and LAMP2A, a lysosomal membrane protein. Recent research has shown that CMA is involved in the maintenance of neuronal protein homeostasis and that reduced CMA activity is involved in the pathogenesis of Parkinson’s disease. The relationship of CMA with neurodegenerative diseases like Parkinson’s has attracted much attention.
Kumamoto University researchers developed an original method for assessing CMA activity in cells and investigated its relationship with the pathogenesis of neurodegenerative diseases. After finding that reduced CMA activity was observed in cells expressing several SCA causing proteins, they hypothesized that reduced CMA activity might be a common part of the pathogenesis of SCA.
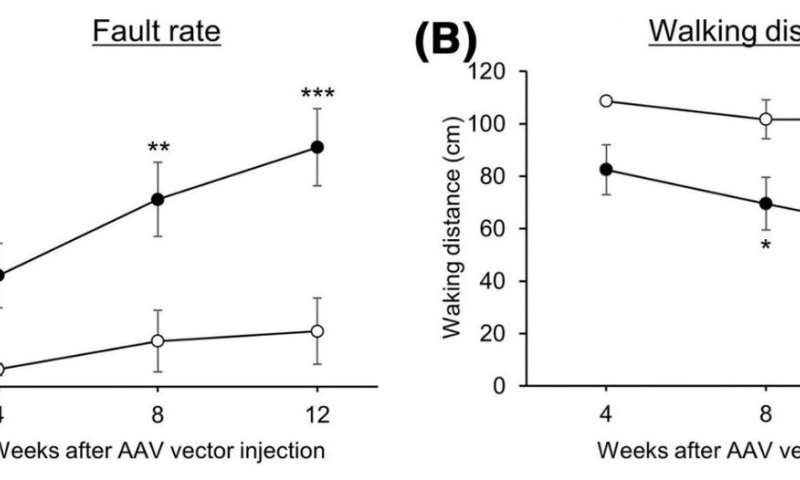
They generated mice with reduced CMA activity in cerebellar neurons by administering adeno-associated viral vectors. These vectors were able to introduce a gene that specifically diminishes LAMP2A into the mouse cerebellum which resulted in progressive motor dysfunction. At early stages of motor dysfunction, histological analysis showed no effect on the morphology of cerebellar neurons or cerebellar structure. However, glial cells, such as astrocytes and microglia, became activated in the cerebellum. Glial activation is a hallmark of neuroinflammation and causes neurodegeneration in various neurodegenerative disorders. At a considerably worse stage of motor dysfunction, glial cell activation as well as cerebellar neurodegeneration and associated atrophy of the cerebellar cortex became prominent.
These effects (progressive motor deficits and neurodegeneration with associated atrophy of the cerebellar cortex) observed in mice with reduced CMA activity in cerebellar neurons are consistent with observations in patients with SCA. Moreover, early glial cell activation has been observed in various SCA mouse models. Since CMA activity has been found to be reduced by the proteins responsible for SCA, the results of this study strongly suggest that reduced CMA activity in cerebellar neurons is a common molecular mechanism in SCA pathogenesis.
Source: Read Full Article