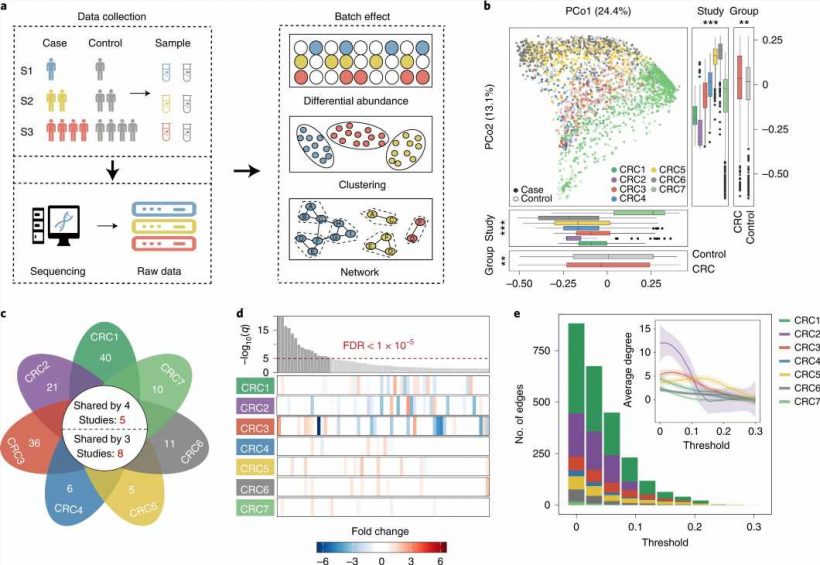
 and CRC samples (filled points) from seven studies. Box plots represent differences among seven studies or between case and control groups. In the box plots: center line, median; box, interquartile range (IQR; the range between the 25th and 75th percentiles); whiskers, 1.5 × IQR; dots, outliers. Two-sided Wilcoxon test or Kruskal–Wallis rank sum test. ***P < 0.001; **P < 0.01; *P < 0.05. c, The number of differentially abundant bacteria with a two-sided Wilcoxon test in each study. The numbers on the leaves correspond to the unique differential bacteria of each study, and differential bacteria shared by multiple studies are shown in the central circle. d, Top: the bar height represents the meta-analysis significance of gut microbial genera derived from blocked Wilcoxon tests (top). Bottom: heatmap representing the fold change within individual studies. Bacteria are ordered by meta-analysis significance. e, The distribution of edges under different thresholds of microbial networks constructed from seven CRC studies. Inset: average degree under different thresholds; different colors of lines represent seven CRC studies, respectively. The gray regions indicate the 95% confidence intervals. Credit: <i>Nature Computational Science</i> (2022). DOI: 10.1038/s43588-022-00247-8")
A research team led by Prof. Zhao Fangqing from the Beijing Institute of Life Sciences of the Chinese Academy of Sciences has proposed a new algorithm (NetMoss) for efficient integration of large-scale microbiome data and biomarker identification.
The study was published in Nature Computational Science on May 23.
The relationship between the gut microbiome and human health has received increasing attention in recent years, and a huge amount of complex data has been accumulated. However, it is challenging to extract information closely related to disease from such big data.
On the one hand, the gut microbiome is more likely to be influenced by factors such as diet and geography. The composition of gut microbiome may vary greatly among different populations, which leads to bias in the direct integration of data and the identification of biomarkers based on abundance. On the other hand, the microbial abundance matrix is too sparse, and it is difficult for conventional computational methods to remove batch effects based on this sparse matrix.
The newly proposed algorithm uses microbial interaction networks to effectively integrate data from different populations. It can quantify the topological differences between different network modules by comparing the perturbations of microbial networks in different states, thus enabling the identification of disease-associated biomarkers.
Compared with previous methods, NetMoss can unbiasedly integrate different batches of microbial data more efficiently, mine disease-associated biomarkers, and identify microbial dysbiosis covariation patterns that drive the occurrence of multiple diseases.
In this study, the researchers collected 11,377 sequencing samples of gut microbiome from diseased and healthy controls, covering 78 studies, 37 diseases, and 13 countries or regions. With these multiple datasets from different populations, they found that currently used computational methods have extreme difficulty removing batch effects caused by experimental and sequencing processes.
To efficiently perform downstream analyses and avoid bias, the researchers developed an efficient computational model for data integration and biomarker identification. The model was based on microbial interaction networks.
Microbial interaction networks are constructed individually and then integrated using different weights based on their structural characteristics. By quantifying the topological differences between different modules in diseased and healthy networks, the bacteria most sensitive to perturbation by external influences are identified as biomarkers.
The researchers applied the computational algorithm to both simulated and real datasets. They found it was highly accurate and robust both in the integrated dataset and in the single dataset.
“Most of the biomarkers did not cause only one disease alone, but were significantly associated with multiple diseases. The similar dysbiosis pattern may provide important clues to the occurrence of different diseases,” said Prof. Zhao.
Source: Read Full Article